

la Drépanocytose
la maladie
|
La drépanocytose (du grec drepanon, faucille), également appelée hémoglobinose S, sicklémie ou anémie à cellules falciformes, est une maladie héréditaire qui se caractérise par l'altération de l'hémoglobine, la protéine assurant le transport de l'oxygène dans le sang. La drépanocytose n'est pas une maladie très rare. Elle est particulièrement fréquente dans les populations d'origine africaine subsaharienne, des Antilles, d'Inde, du Moyen-Orient et du bassin méditerranéen particulièrement en Grèce et en Italie. On estime que 50 millions d'individus en sont atteints dans le monde. C'est la première maladie génétique en France, et probablement dans le monde. HistoriqueEn 1904, James Herrick, médecin à Chicago, fait la première description médicale de la drépanocytose : il examine un étudiant noir âgé de 20 ans, hospitalisé pour toux et fièvre. Le sujet est faible, a des vertiges et souffre de maux de tête. Depuis un an, il ressent des palpitations et un essoufflement comme certains membres de sa famille. L’examen du sangmontre que le malade est très anémique, le nombre de ses hématies n’atteignant que la moitié de la valeur normale. L’observation d’un frottis sanguin montre des hématies inhabituelles en forme de faucille ou feuille d'acanthe. En 1949, James Neel démontre que la transmission de cette maladie est mendélienne. La même année Linus Paulingmontre qu'elle est due à une structure anormale de l'hémoglobine, caractérisée par une moindre solubilité. C'est ainsi la première fois qu'on découvre l'origine moléculaire d'une maladie génétique. En 1956, le Britannique Vernon Ingram montre qu'elle est due à un remplacement d'un acide aminé dans l'hémoglobine anormale. Cela a démontré pour la première fois que lesgènes déterminaient la nature de chaque acide aminé dans une protéine. En 1978, Tom Maniatis isole le gène de la bêta globine. En 1980, Yuet Wai Kan (en) met au point un test génétique prénatal de la drépanocytose. PathogénieAu niveau cellulaireLes globules rouges de l'homozygote ne contiennent pratiquement que de l'HbS. Or, ces molécules ont la propriété de se polymériser lorsqu'elles sont désoxygénées, donnant lieu à la formation de fibres qui déforment le globule et lui donne un aspect en faucille. Ceci explique que la falciformation des hématies soit déclenchée par le manque d'oxygène dans le sang (hypoxie) :
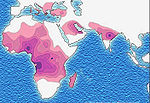
Au niveau moléculaireL'allèle S est un allèle anormal du gène régissant la structure de la chaîne bêta de l'hémoglobine. Il est responsable de la synthèse de chaînes bêta, dont un acide glutamique en position 6 est remplacé par une valine. L'hémoglobine qui en résulte appelée HbS (hémoglobine S pour Sickle-cell disease, dénomination anglaise de la maladie) a donc la structure "alpha2ß2S". Elle se distingue de l'hémoglobine A, normale, par sa mobilité électrophorétique plus lente, mais surtout par l'insolubilité de sa forme désoxygénée, quicristallise facilement. Cela forme des fibres longues qui vont déformer le globule rouge. Au niveau génétiqueC'est le gène qui code la chaîne Béta de l'hémoglobine qui est impliqué. Ce gène est porté par le chromosome 11. La version mutée (l'allèleS) de ce gène est responsable de l'anémie falciforme. C'est un allèle co-dominant (codominance). C'est-à-dire que lorsque l'on est hétérozygote, les deux allèles s'expriment, cela pour la simple raison que chaque allèle code pour une chaîne de Beta-globine. Cependant chez cet individu, on ne constatera pas de symptômes, tout en possédant des hématies falciformes (on pourrait la considerer comme une forme atténuée). Cette forme est appelée (S/A). La forme homozygote est la (S/S). Il s'agit de la plus douloureuse, celle dans laquelle sont décrites les crises. Chez l'hétérozygote (S/A), les globules rouges ne contiennent pas nécessairement un mélange en proportions égales d'HbA et d'HbS. Au niveau de l'organismeLes globules rouges ayant perdu leur élasticité vont obstruer les capillaires provoquant une ischémie par manque d'apport d'oxygène au niveau de différents territoires. Cela explique les crises douloureuses (infarctus osseux), les infarctus cérébraux, etc. Les globules rouges pourraient également léser la paroi interne des vaisseaux (endothélium) entraînant un risque d'obstruction de ces derniers. Ces globules rouges sont plus fragiles et vont se rompre beaucoup plus facilement, expliquant l'anémie de type hémolytique (par destruction hématies). ÉpidémiologieSelon l'OMS, 300 000 enfants naissent dans le monde chaque année avec une anomalie majeure de l'hémoglobine dont la plus fréquente est celle de la drépanocytose. En France métropolitaine, 12 000 personnes seraient atteintes d'un syndrome drépanocytaire majeur, 2 000 en Martinique et 1 500 enGuadeloupe. L'allèle S, responsable de l'anomalie, est surtout répandu dans le continent africain (atteignant dans certaines populations la fréquence de 30 %); on le trouve également en Inde, en Arabie saoudite et dans d'autres régions du bord de la Méditerranée, en Italie (surtout en Sicile), en Grèce et enAnatolie. Les migrations ont accru la fréquence de cette maladie sur le continent américain. Cette distribution se superpose assez bien avec celle d'une autre maladie, le paludisme ou malaria, qui, elle, a une origine infectieuse: lePlasmodium falciparum. La présence élevée de cette maladie en Afrique semble être un cas de polymorphisme génétique équilibré entraîné par une sélection naturelle (voir :avantage hétérozygote) : en effet, les personnes porteuses saines hétérozygotes (A/S) ou atteintes de la drépanocytose homozygotes (S/S) sont protégées des affections neurologiques de Plasmodium, le parasite responsable du paludisme aussi appelé malaria.
AfriqueDans certaines parties de l’Afrique subsaharienne, la drépanocytose touche jusqu’à 2 % des nouveau-nés. La fréquence du trait drépanocytaire, c'est-à-dire le pourcentage de porteurs sains qui n’ont hérité du gène mutant que d’un seul des parents, atteint 10 à 40 % en Afrique équatoriale, 1 à 2 % sur la côte de l’Afrique du Nord et moins de 1 % en Afrique du Sud. Dans les pays d’Afrique de l’Ouest (Ghana et Nigéria), la fréquence du trait drépanocytaire atteint 15 à 30 %. En Ouganda, cette fréquence atteint 45 % chez les Baambas. EuropeDans plusieurs pays ou régions d'Europe (Sud de l'Italie, Grèce, Sud du Portugal, Albanie, Sud de la Turquie), la drépanocytose est indigène avec des fréquences de porteurs du trait entre 1 et 5 % de la population. Dans d’autres pays européens (Royaume-Uni, France, Belgique, Allemagne) la maladie est apparue avec les flux migratoires en provenance d'Afrique, du Moyen-Orient et d'Asie. SymptômesL'affection se signale chez le nourrisson, mais n'est d'ordinaire pas manifeste à la naissance parce que les globules rouges du nouveau-né contiennent encore 50-90 % d'hémoglobine fœtale. Les symptômes de cette maladie peuvent apparaître dès l'âge de deux à trois mois, date d'apparition de la chaîne Béta. Les manifestations aiguës habituelles de la drépanocytose sont de trois ordres :
Les manifestations chroniques de la drépanocytose associent un retard de taille et de poids, des déficits nutritionnels (en folates, car cette vitamine est indispensable à la création des hématies qui sont renouvelées très rapidement lors des crises d'anémie, épuisant ainsi le stock de folates), un retard pubertaire fréquent, des troubles cardio-pulmonaires (augmentation de la taille du cœur, insuffisance respiratoire), une rate augmentée de volume ou atrophiée, des anomalies rétiniennes (hémorragies), etc. 
Image prise au microscope électronique du sang d'un patient hétérozygote pour la drépanocytose, sur laquelle on distingue des globules rougessains, circulaires et concaves, des globules rouges malades possédant de l'hémoglobine mutée S, de la forme d'une faucille. Examens et diagnosticÀ part les constatations communes à toutes les anémies hémolytiques, le diagnostic repose sur la mise en évidence de l'hémoglobine S. Ceci peut se faire :
Dans les pays industrialisés, le diagnostic se fait en période néo-natale si les parents sont à risque ou atteints. Dans les pays non industrialisés, le diagnostic se fait souvent à la première manifestation ou complication. Le dépistage néo natal pourrait se traduire par une amélioration du pronostic. Atteinte oculaireL'atteinte oculaire est plus fréquente et plus grave chez les patients SC par rapport aux patients SS. Un examen régulier est nécessaire afin de dépister précocemment la rétinopathie drépanocytaire afin de pouvoir proposer un traitement. PronosticLa maladie peut se compliquer d'infarctus cérébraux dont la prévalence atteint près de 10 % des patients âgés de moins de 20 ans. L'hypertension artérielle pulmonaire est une complication fréquente et grave. Elle est retrouvée en échocardiographie dans près d'un tiers des cas mais cet examen s'avère peu spécifique et la prévalence réelle (confirmée par la mesure directe de la pression pulmonaire par cathétérisme cardiaque) serait bien moindre. Avant l'apparition de l'hydroxyurée près de la moitié des malades décédait avant l'âge de 50 ans, soit au cours d'une crise, ou d'un accident vasculaire cérébral. TraitementsLe traitement de la drépanocytose repose sur :
L'hydroxyurée permet de favoriser la production d'hémoglobine fœtale, formée habituellement en petite quantité et parfaitement fonctionnelle, en inhibant la production d'hématies contenant l'hémoglobine S. Ce médicament semble réduire significativement le nombre de crises douloureuses et la mortalité de la maladie. Il ne peut cependant pas être utilisé, de par son mécanisme d'action, chez les patients anémiques. La surveillance des paramètres sanguins doit donc être très soigneuse. L'autre obstacle à son utilisation reste son coût, qui peut toutefois générer des économies de prise en charge dans les pays économiquement développés. La prévention des infections par le pneumocoque chez le jeune enfant est faite par la vaccination. Des transfusions sanguines pourraient diminuer sensiblement le risque d'accidents vasculaire-cérébraux chez certains enfants particulièrement à risque (anomalie du doppler trans-crânien). Mesures simples de préventionPour éviter les crises il est recommandé de suivre des mesures simples suivantes:
Greffe de moelle osseuseLes hématies sont produites à partir de cellules souches dans la moelle osseuse. En détruisant la moelle osseuse du malade et en la remplaçant par celle d'un donneur, il y a possibilité d'obtenir une guérison totale. Environ 200 greffes ont été réalisées dans le monde chez des drépanocytaires, permettant d'obtenir la guérison dans 85 % des cas. Il faut cependant un donneur apparenté le plus possible: un frère ou une sœur. Il y a la possibilité pour les parents de recourir à une fécondation in vitro avec sélection par DPI d'embryons compatibles pour la greffe. Cette voie de traitement dite du « bébé médicament » est très encadrée par les lois de bioéthique. Article connexe : Transplantation de moelle osseuse.
Voies de rechercheDes souris drépanocytaires ont pu être guéries en introduisant chez ces animaux un gène produisant une hémoglobine "anti-drépanocytaire" en quantité élevée. En 2007, le VK500 est proposé dans le traitement de la drépanocytose. Cependant, aucune étude clinique sérieuse n'a prouvé l'efficacité réelle de ce médicament. source: http://fr.wikipedia.org/wiki/Dr%C3%A9panocytose |
Commentaires
-
 Thankfulness to my father who stated to me regarding this website, this weblog is genuinely awesome.
Thankfulness to my father who stated to me regarding this website, this weblog is genuinely awesome. -
 GRAND MAITRE SPIRITUEL voyant-koffi
GRAND MAITRE SPIRITUEL voyant-koffi
TEL (00229)67655902 (+229)67655902
Email : voyant-koffi.akpota@outlook.com
JE SUIS DISPONIBLE SUR WHATSAPP SI VOUS AVEZ BESOIN D'AIDE
solutions immédiates a tous vos problèmes spirituels
Retour amour - Retrouvez l'être aimé en 7 Jours
ENVOÛTEMENT D'AMOUR RETOUR D'AFFECTION
JE PRATIQUE DES RITUELS POUR:
RETOUR D'AFFECTION
AMOUR
ARGENT
VENTE DE BIENS
REUSSITE AUX EXAMENS
PERMIS DE CONDUIRE
AMELIORATION DU CHIFFRE D AFFAIRE
APPORT DE CLIENTÈLE
SÉPARATION
TEL (00229)67655902 (+229)67655902
Émail :voyant-koffi.akpota@outlook.com -
 I constantly emailed this website post page to all my associates, since if like to read it then my links will too.
I constantly emailed this website post page to all my associates, since if like to read it then my links will too. -
 I used to be suggested this blog by way of my cousin. I am no longer sure whether or not this publish is written via him as no one else recognise such detailed about my trouble. You are amazing! Thank you!
I used to be suggested this blog by way of my cousin. I am no longer sure whether or not this publish is written via him as no one else recognise such detailed about my trouble. You are amazing! Thank you! -
 My brother suggested I would possibly like this blog. He was entirely right. This submit actually made my day. You cann't consider just how much time I had spent for this info! Thank you!
My brother suggested I would possibly like this blog. He was entirely right. This submit actually made my day. You cann't consider just how much time I had spent for this info! Thank you! -
 Incredible quest there. What happened after? Take care!
Incredible quest there. What happened after? Take care! -
 Outstanding story there. What happened after? Thanks!
Outstanding story there. What happened after? Thanks! -
 An interesting discussion is worth comment. There's no doubt that that you need to write more about this topic, it may not be a taboo matter but generally folks don't talk about these issues. To the next! Many thanks!!
An interesting discussion is worth comment. There's no doubt that that you need to write more about this topic, it may not be a taboo matter but generally folks don't talk about these issues. To the next! Many thanks!!
Ajouter un commentaire